Mutation Mapping Pipiline for C. elegans EMS mutagenesis and Backcross Experiments
Richard J. Acton
2025-12-22
Generating-input.RmdQuick Start
-
Inputs:
- paired-end fastq files to a galaxy (Afgan et al. 2016; Jalili et al. 2020) history as
list of dataset pairs - A suitable genome fasta file (C. elegans, ce11.fa.gz - Compatible with WBcel235.86 used by SnpEff)
- paired-end fastq files to a galaxy (Afgan et al. 2016; Jalili et al. 2020) history as
- Run the Pipeline: https://usegalaxy.eu/u/richardjacton/w/c-elegans-ems-mutagenesis-mutation-caller
-
Outputs:
-
MultiQCHTML report with QC info on the input fastqs, trimming, mapping, and deduplication steps. -
.vcffile with variants from all samples (FreeBayes mutation caller) -
.vcffile with variants from all samples (MiModD mutation caller) -
.gfffile with deletions from all samples (MiModD deletion calling tool)
-
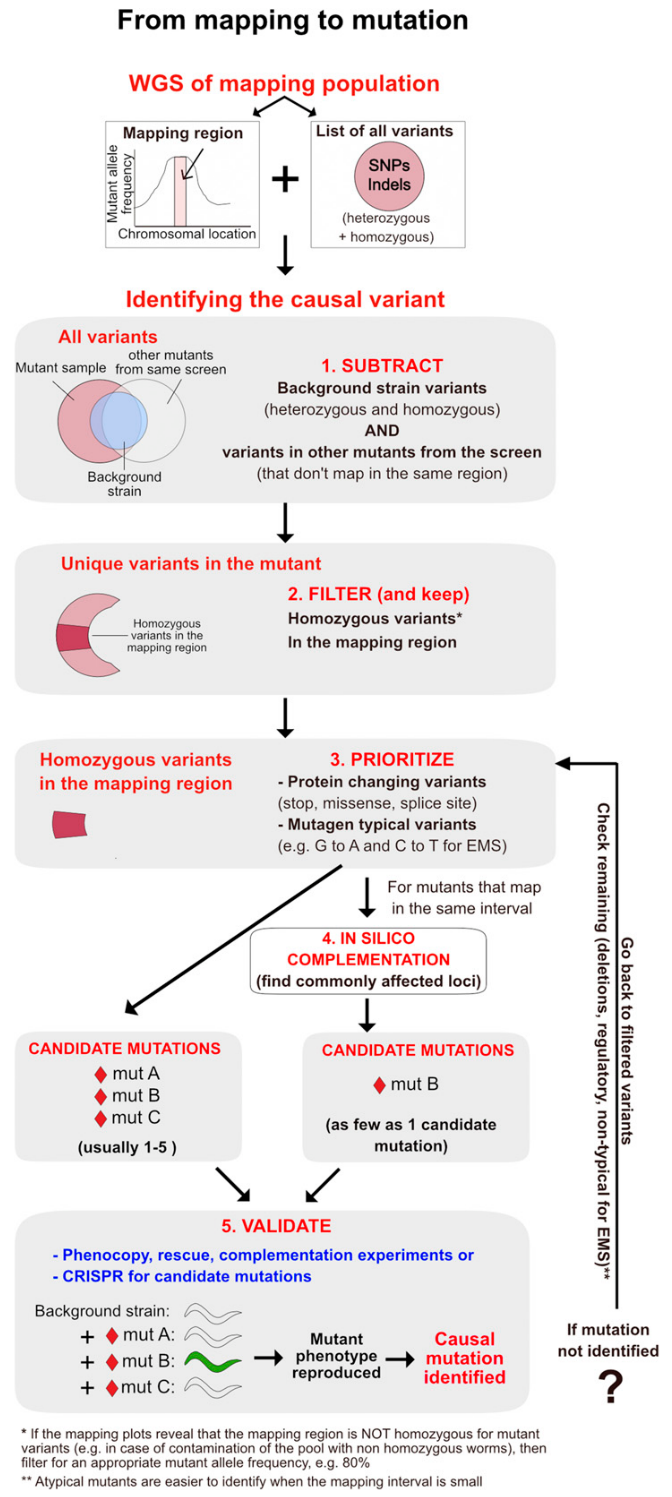
- Perform Quality filtering and appropriate set subtractions with
MutantSetsor alternatively theMiModD VCF FilterorSnpSift Filtertools to identify candidate variants. - (optionally)
MiModD NacreousMapfor visualisation of mutation locations andMiModD Report Variantsfor HTML mutation list
NB samples are expected to be of the form
‘A123_0001_S1_R1_L001.fq.gz’, sample Identifiers are extracted from this
with a regular expression: \w+_(\d+)_S\d+_L\d+.*. This
would yield the sample identifier of: 0001. If your file does not
conform to this pattern you may need to update this regex by editing the
rules in the ‘apply rule to collection’ step of the workflow.
Background
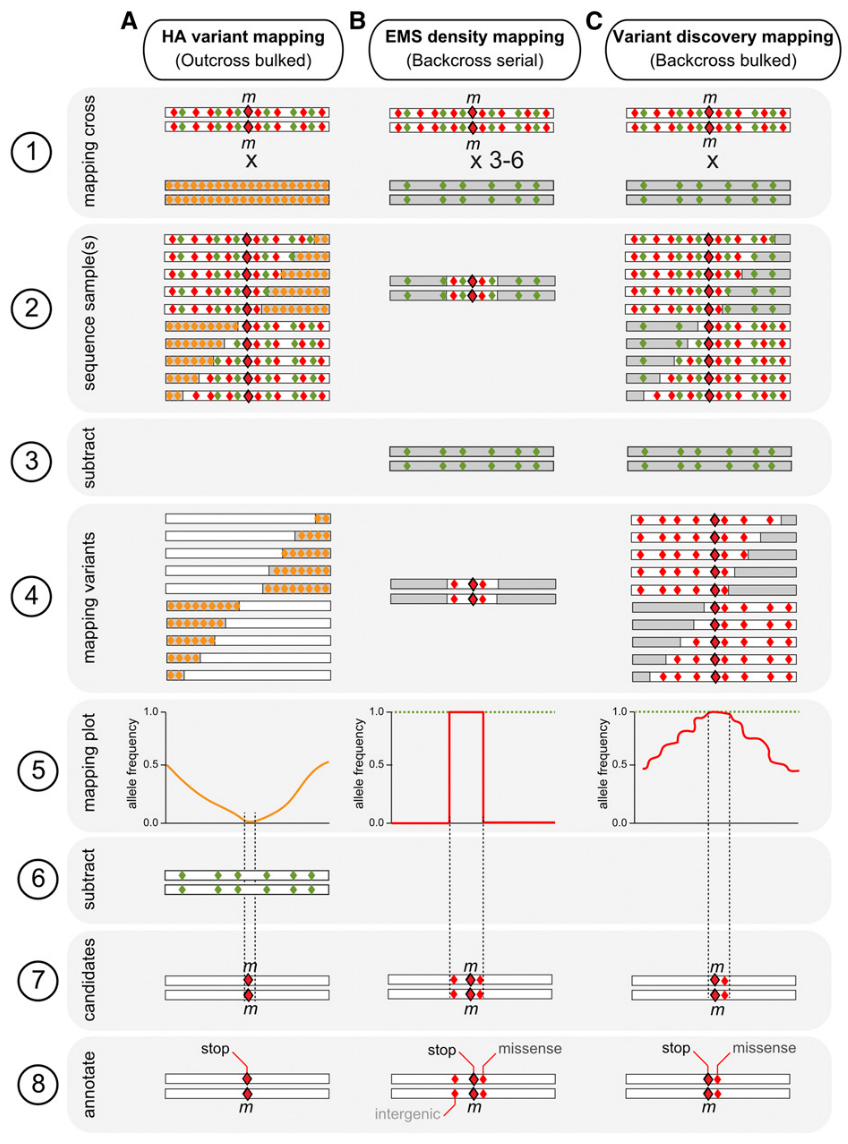
Doitsidou et al. reviewed Sequencing-Based Approaches for Mutation Mapping and Identification in C. elegans (Doitsidou, Jarriault, and Poole 2016). They describe three main approaches to mapping by sequencing:
- Hawaiian variant mapping
- EMS-density mapping
- Variant discovery mapping
This pipeline is currently only compatible with 2 of them, EMS-density mapping & Variant discovery mapping (VDM).
Research Need
The Schumacher lab identified a need for an analysis pipeline to map and identify mutations in Ethyl methanesulfonate (EMS) mutagenesis forward genetic screens.
Previously a tool called CloudMap (Minevich et al.
2012) had been used for this purpose on a Galaxy server.
CloundMap is no longer under active development and has
been deprecated from Galaxy Europe
and replaced by MiModD Docs
Choice of Tools
In a comparison of C. elegans mutation calling pipelines
Smith et al. (Smith and
Yun 2017) indicated that they had good results with the
FreeBayes (Garrison and Marth 2012). So I have
initially included this tool here in addition to theMiModD
mutation caller to evaluate their relative performance. They also found
the the BBMap aligner yielded better results however this
is not available in Galaxy so I have opted for Bowtie2 for
expediency.
Pipeline Summary
https://usegalaxy.eu/u/richardjacton/w/c-elegans-ems-mutagenesis-mutation-caller
- Adapter and Quality Trimming with
fastp(Chen et al. 2018) - Alignment with
bowtie2 --sensitive-local(Langmead and Salzberg 2012) -
samtools viewrequiring that reads are mapped in a proper pair (Li et al. 2009) - Removal of PCR duplicates with
Picard MarkDuplicates(“Picard Toolkit” 2019) - Left alignment of indels in the BAM files using
FreeBayes(Garrison and Marth 2012) -
MultiQCaggregating quality metrics from trimming, deduplication and alignment (Ewels et al. 2016) - Variant calling with
FreeBayes(Garrison and Marth 2012),MiModD(“MiModD” 2013) variant caller and deletion caller - SNP effect annotation with
SnfEff eff(Cingolani et al. 2012) - SNP type annotation with
SnpSift Variant Type(Cingolani et al. 2012)
Instructions (Step-by-Step)
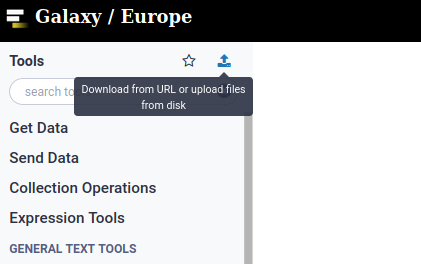
1. Upload Data to galaxy
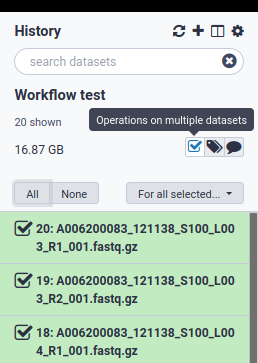
2. Select all fastq files and create a paired list
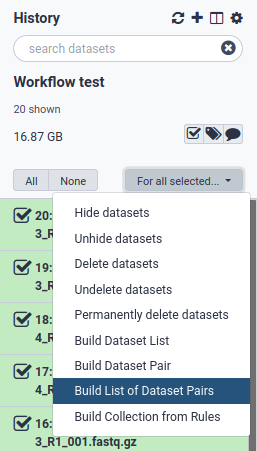
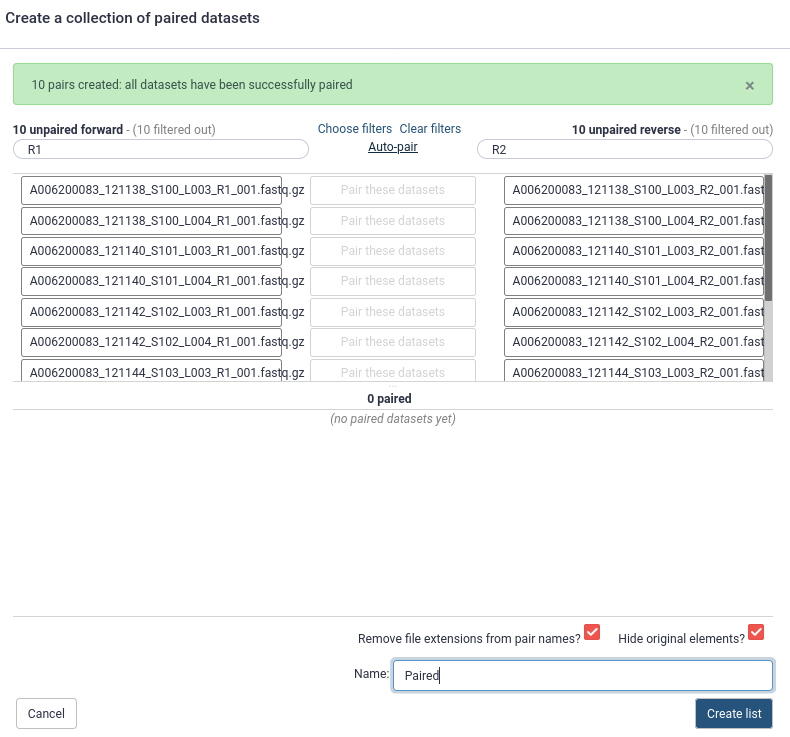
3. Pair the fastq files
4. Import the workflow
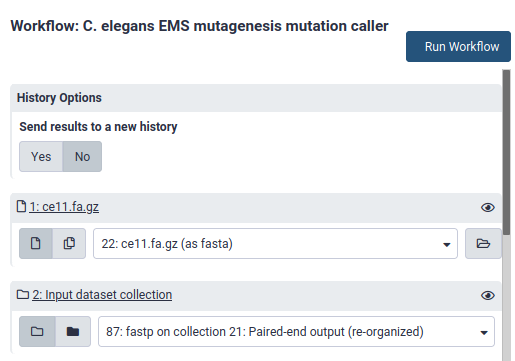
5. Run the workflow
Select the paired list object and a genome sequence file as inputs
6. Check Quality Control Information
Inspect the MultiQC output for signs of technical
problems with your data. Consult with your friendly local
bioinformatician if there are QC issues you can’t diagnose.

7. Preliminary quality filtering
SnpSift filter
Locate the SnpSift filter
tool in the galaxy tools panel and apply some initial quality filters,
simply ( QUAL > 15) or 20 is probably
sufficient. Starting with a low stringency filter and applying more
stringent criteria when inspecting your candidate mutations it is
probably advisable to avoid throwing out possible mutations. Some
initial filtering is advisable as the full-sized VCF files may be too
large to be easily read by the candidate mutant inspection tool in the
next steps. You can check how many lines are in your VCF files by
selecting them in the Galaxy history.
8. Download Data
The main FreeBayes VCF file:
The MiModD deletion calls:
9. Load the results in the MutantSets Shiny App
to identify candidate mutations
If running the App locally, install the R package from: https://github.com/RichardJActon/MutantSets
R package installation and running the app locally:
# install.packages("remotes") # If you don't already have remotes/devtools
# remotes::install_github("knausb/vcfR") # If vcfR fails to install from CRAN
remotes::install_github("RichardJActon/MutantSets")
MutantSets::launchApp() # opens the app in a web browser- Load the VCF and (optionally) the gff deletion mutant files into
MutantSets - (Optionally) Name your samples something easier to understand
- Use the genotype filters to subtract the appropriate sets
- Tweak quality and allele frequency thresholds to get a small set of high quality candidates
- Assess the candidate mutations by clicking on them and looking at their predicted effects and genomic locations
- Download your top results as a
.tsvfile (openable in excel)
You should now have some candidate mutants to screen - Good Luck!
Feedback
Please direct bug reports, feature requests, and questions to the maintainer of the mutant sets package via [github issues](https://github.com/RichardJActon/MutantSets/issues.